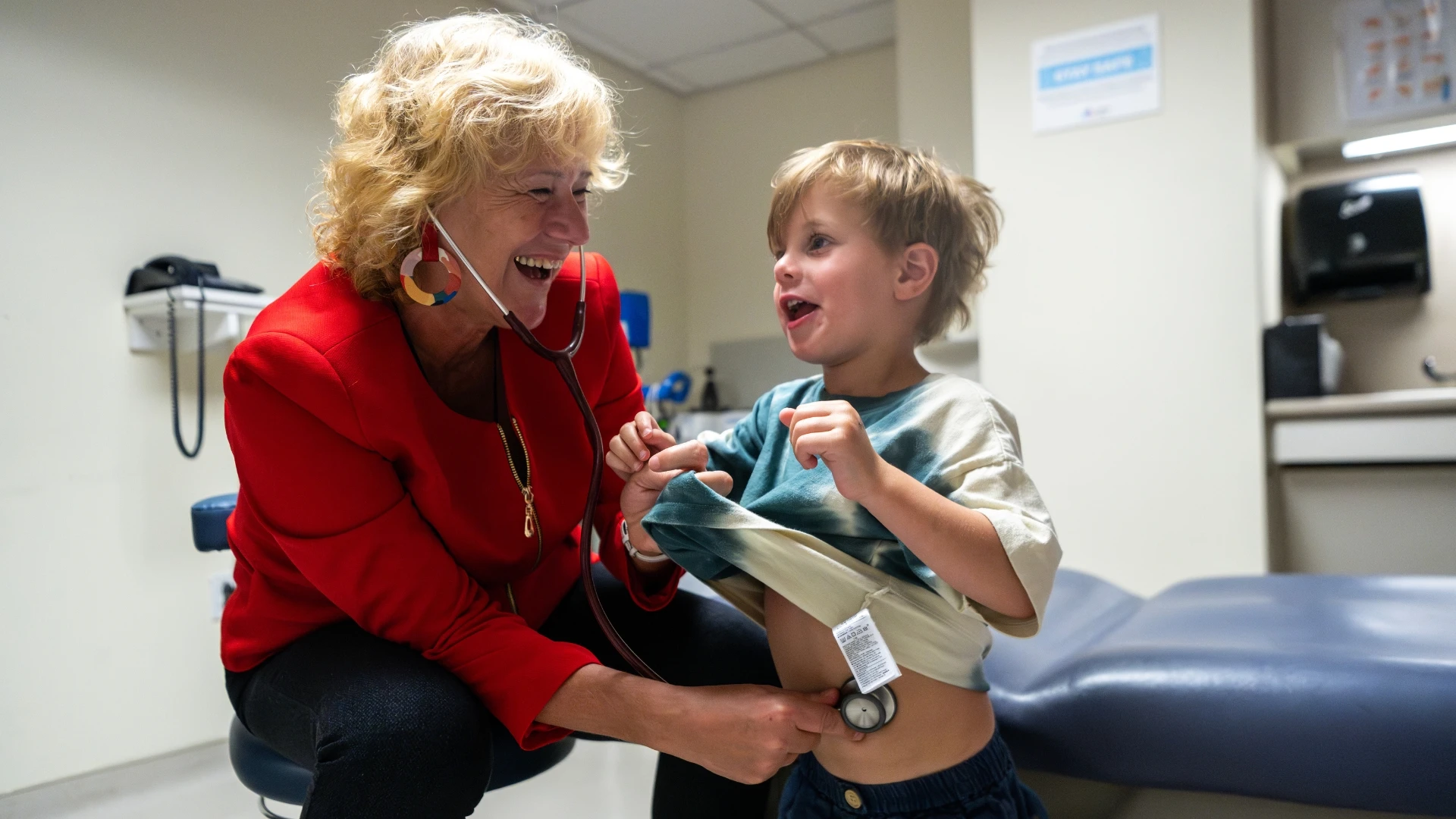
Over their decades of experience, Eva Morava and Tamas Kozicz have each funneled their individual expertise — hers in metabolic disorders and clinical research, his in neuroscience and organ models — into a shared mission to treat rare diseases. Originally from Hungary, the husband-and-wife team spent time at the Mayo Clinic before joining Mount Sinai’s Department of Genetics and Genomic Sciences as senior faculty in February 2024.
“Our joint lab focuses on rare diseases — and we can even call them ultra-rare disorders, because some of the patient cohorts we work with include only ten individuals in the United States,” says Kozicz, MD, PhD, Professor of Genetics and Genomic Sciences and Neuroscience at the Icahn School of Medicine at Mount Sinai. “That, of course, has its challenges, but it’s also a fantastic opportunity to understand these really understudied patient populations.”
Their research aims to develop novel therapies for a group of genetic disorders called inborn errors of metabolism, which affect 1 in 2,500 births. They are caused by pathogenic variants in genes coding for proteins that function in metabolism. There are thousands of different diseases in total, but Morava and Kozicz specialize in two subgroups of inborn errors of metabolism known as congenital disorders of glycosylation (CDG) and primary mitochondrial disorders (PMD).
CDG include over 190 rare metabolic disorders that arise due to defects in glycosylation, the process of attaching sugar molecules to proteins or lipids. CDG are usually apparent from infancy and can affect any part of the body, usually with a significant neurological component. The disorders are heterogeneous and can vary in severity, from relatively mild to severe, disabling, or life-threatening.
PMD, on the other hand, are caused by defects in mitochondria, the power plants of the cell. An estimated 1 in 5,000 people has a genetic mitochondrial disease, whose symptoms can include muscular and neurological problems, along with impaired vision, hearing loss, cardiac arrhythmia, diabetes, and stunted growth.
“At the Mayo Clinic, we established our shared lab and aligned our research interests to work on central nervous system aspects of PMD and CDG,” says Morava, MD, PhD, Professor of Genetics and Genomic Sciences and Pediatrics at Icahn Mount Sinai. “From discovering new disorders, we actually turned our interest towards developing new therapies within the framework of individualized, personalized medicine.”


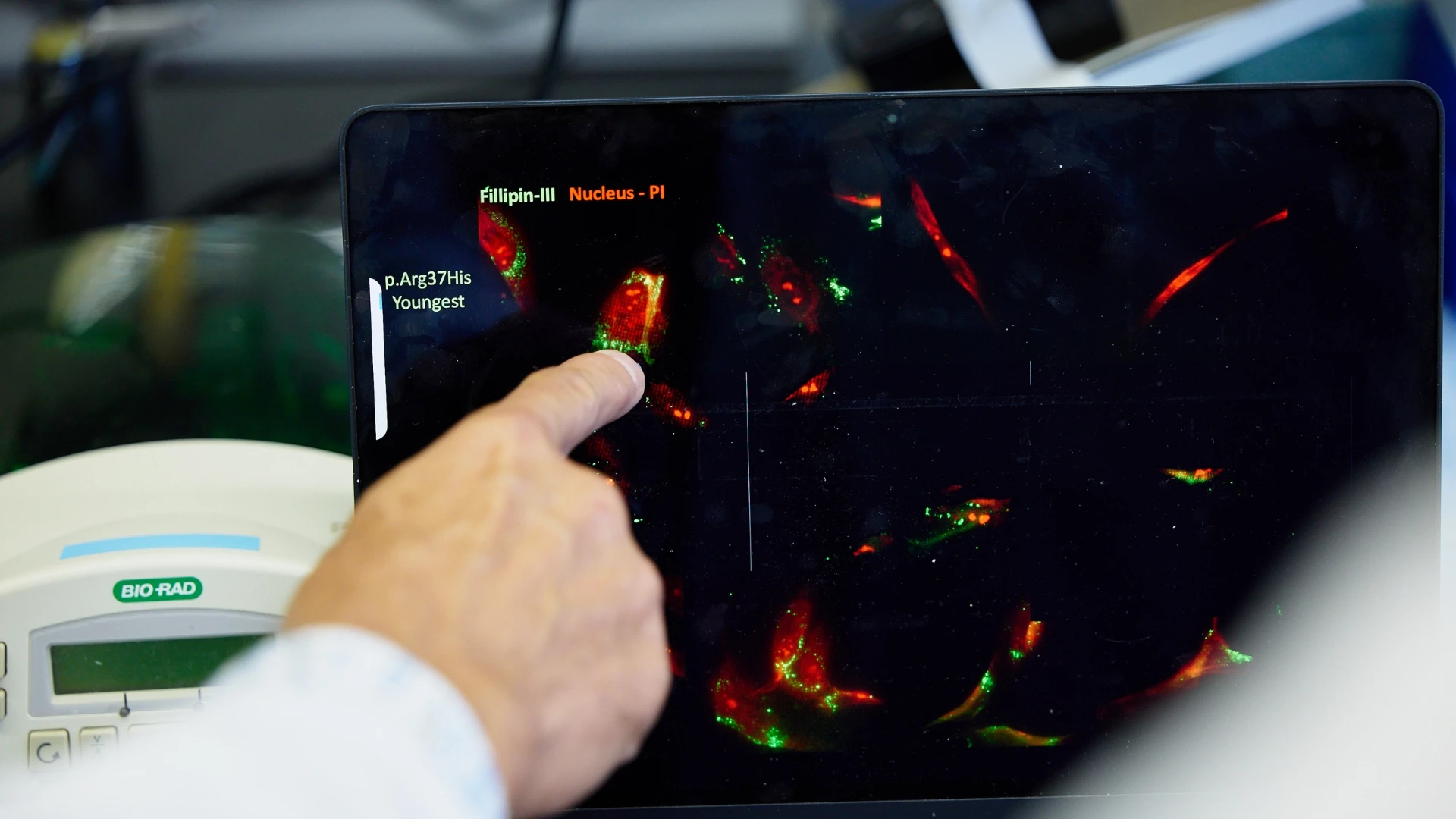
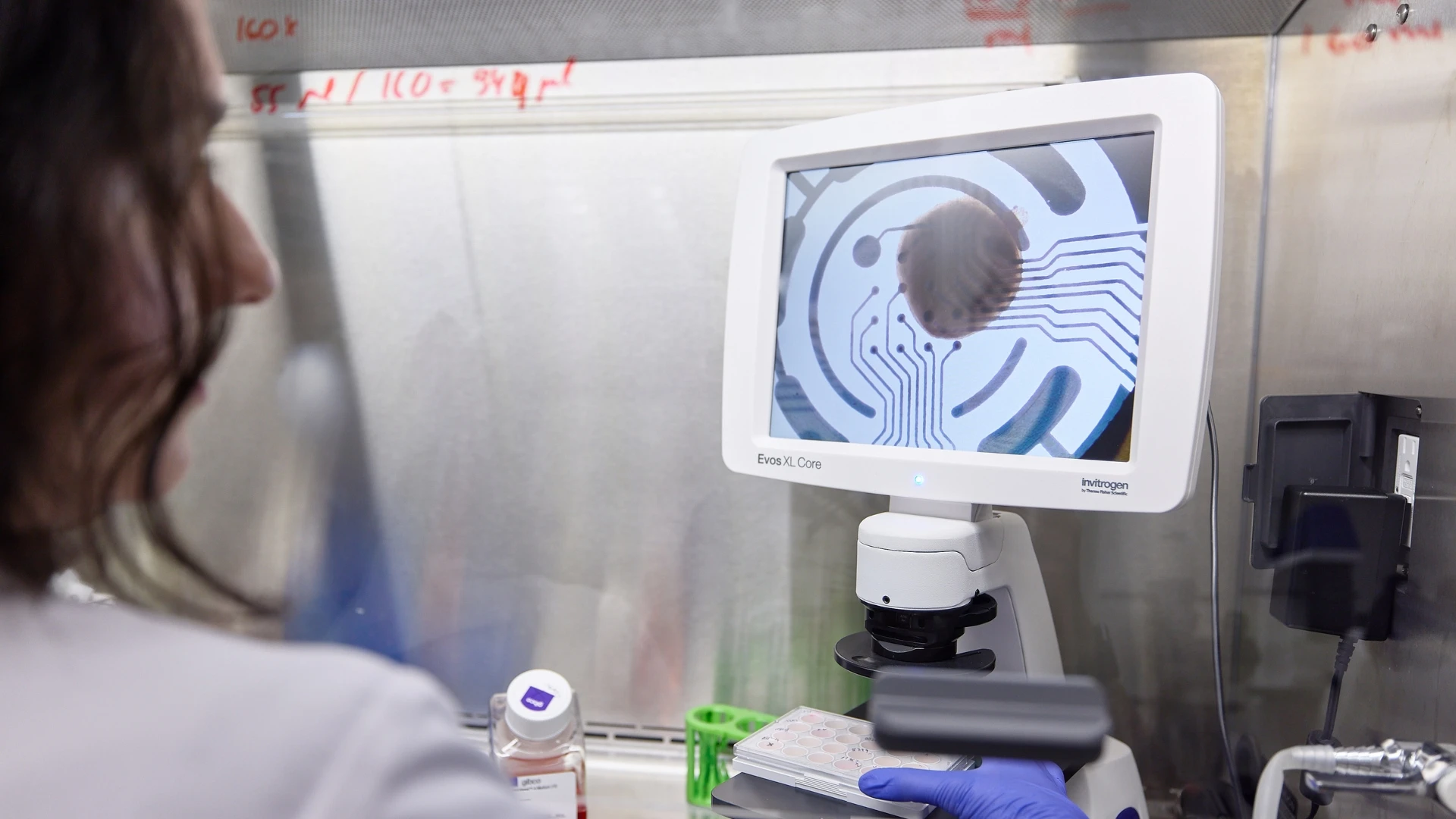
In recent years, their lab has developed organ models for several rare disorders, identified novel drug targets, and successfully translated potential therapies to clinical trials in humans.
Previously, Kozicz designed a pipeline for creating 3D neural cultures, known as human cortical organoids, using a method that turns patient skin cells into pluripotent stem cells. Such organ models provide powerful tools to investigate human brain development and neurodevelopmental disorders. He has also created organoids for the heart, lung, and liver with collaborators at Mount Sinai.
“Using these patient skin cells, we can develop different tissues which are not just sharing the patient’s genetic background and disease-causing variants, but they are also organ-specific,” he says. “Many of the disorders that we study have specific organ presentations, and now we can really use these in vitro models to look at issues in a disease-relevant context.”
Kozicz and his colleagues employ these model systems — which capture cell type complexity and species specificity better than animal models or traditional cell cultures — to find novel treatments for CDG and PMD. Patient-derived organoids can be used to predict patient-specific responses to drugs, meaning they can more efficiently screen multiple drugs to find possible candidates. Then, the researchers go back to the organoids to test each candidate and its mechanisms of action more thoroughly.
“Once the circle is completed, and we identify potential drugs, then Eva becomes really involved in bringing those into a clinical trial,” Kozicz says. “Our ultimate goal is always to bring treatments into clinical trials and make a new drug available for individuals with these really devastating and, up until now, basically untreatable disorders.”
Morava serves as the Principal Investigator for the Frontiers in Congenital Disorders of Glycosylation Consortium (FCDGC), a National Institutes of Health (NIH)-Funded Rare Diseases Clinical Research Network Consortium dedicated to studying CDG. At Mount Sinai, Morava will soon launch pivotal first-in-human trials in two types of CDG, known as PGM1-CDG and SLC35A2-CDG. The Phase II studies are designed to assess the efficacy, safety, and tolerability of an ultra-pure formulation of galactose, a naturally occurring simple sugar found in dairy products and fruit.
Their earlier results suggest that daily oral supplementation with galactose may benefit certain individuals with CDG. Out of five patients with PGM1-CDG, four experienced notable clinical improvement after supplementation. Similarly, a first pilot study of galactose therapy involving ten patients with SLC35A2-CDG demonstrated safety and efficacy of the treatment.
“In these disorders, a unique sugar derived from milk seems to have a major impact on the patient’s clinical outcome, with improvement in liver function, coagulation, thrombosis risk, and even seizure frequency,” says Morava. “So, it’s a very exciting approach that uses nutritional interventions to treat such severe symptoms.”
The researchers also plan to start a clinical trial at Mount Sinai for PMM2-CDG, a type of CDG that affects more than 1,000 individuals worldwide, based on their recent organoid-based laboratory experiments.
“I feel that my patients are my second family, and to be able to translate bench research to clinical trials and have patients actually benefit from these findings is a mind-blowing experience,” she says. “It’s really fulfilling.”