The Cardiovascular Research Institute (CVRI) in 2024 continued to advance the understanding of the heart and blood in a broad physiological context, under the leadership of Director Filip K. Swirski, PhD.
“The cardiovascular system is part of an intricate network that involves many tissue, cellular, and molecular processes. The immune, nervous, metabolic, and hematopoietic systems, just to name a few, all play a role in our cardiovascular health,” Dr. Swirski says. “Understanding the mechanisms of inter-system communication is key to innovation in fundamental and translational science.
Among significant achievements, a team led by Jason C. Kovacic, MD, PhD, and Jeffrey W. Olin, DO, conducted a systems biology study that identified a gene-regulatory network that drives fibromuscular dysplasia, suggesting pathways for future therapies for this challenging and poorly understood disease.
In another groundbreaking study, a Mount Sinai team was the first to demonstrate how the heart and brain communicate with each other through the immune system to promote sleep and recovery after myocardial infarction (MI). The study found that in humans and mice, monocytes are actively recruited to the brain after MI to augment sleep, which suppresses sympathetic outflow to the heart, limiting inflammation and promoting healing—a finding that suggests sleep should be a crucial focus in clinical management after cardiac events. Cameron McAlpine, PhD, was senior author of the study, and Dr. Swirski was among the co-authors.
A study led by Carol Gregorio, PhD, discovered mutations that lead to dilated cardiomyopathy in a finding that could have therapeutic potential. Lior Zangi, PhD, and team identified the Lin28a gene as an important regulator of the heart muscle cells’ proliferation, improving cardiac function post-heart attack in a mouse model.
The investigators provide some insights into their work below.
Zangi Lab

Lior Zangi, PhD, center, with from left, Rachel Hanan, Lucia Zigova, Matthew E. Adjmi, Matteo Ghiringhelli, DVM, PhD, Segev Sharon, Eftychia Markopoulou, and Gayatari Mainkar.
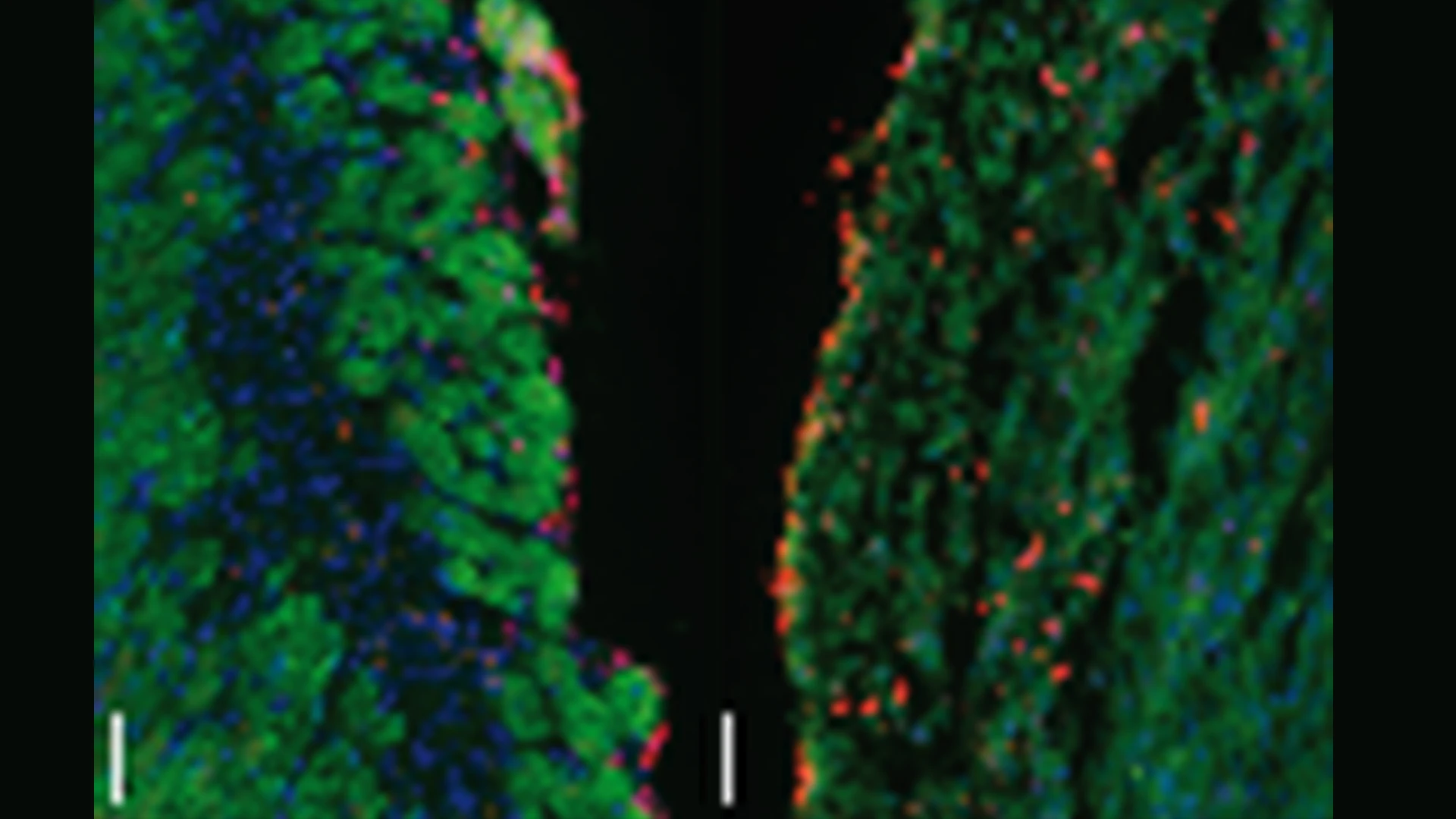
Lin28a cardiomyocyte-specific modRNA translational system (CM SMRT) substantially decreased the death of CMs compared to control seven days after MI. This suggests Lin28a has an additional cardio-protective function beyond its effect on cardiac regeneration post-MI.
Abstract of Study: Lin28a cardiomyocyte-specific modified mRNA translation system induces cardiomyocyte cell division and cardiac repair, Journal of Molecular and Cellular Cardiology, March 2024
The mammalian heart has a limited regenerative capacity. Previous work suggested the heart can regenerate during development and immediately after birth by inducing cardiomyocyte (CM) proliferation; however, this capacity is lost seven days after birth. ModRNA gene delivery, the same technology used successfully in the two mRNA vaccines against SARS-CoV-2, can prompt cardiac regeneration, cardiovascular regeneration, and cardiac protection. We recently established a novel CM-specific modRNA translational system (SMRT) that allows modRNA translation only in CMs. We demonstrated that this system delivers potent intracellular genes (e.g., cell cycle-promoting Pkm2), which are beneficial when expressed in one cell type (i.e., CMs) but not others (non-CMs). Here, we identify Lin28a as an important regulator of the CM cell cycle. We show that Lin28a is expressed in CMs during development and immediately after birth, but not during adulthood. We describe that specific delivery of Lin28a into CM, using CM SMRTs, enables CM cell division and proliferation. Further, we determine that this proliferation leads to cardiac repair and better outcome post-MI. Moreover, we identify the molecular pathway of Lin28a in CMs. We also demonstrate that Lin28a suppresses Let-7, which is vital for CM proliferation, partially due to its suppressive role on cMYC, HMGA2, and K-RAS.
Notes From the Investigator: Lior Zangi, PhD, Associate Professor of Medicine (Cardiology), and Genetics and Genomic Sciences
Heart muscle cells are unable to proliferate after birth. Therefore, after MI, the heart is unable to create newly formed muscle, and the heart remains scarred, with reduced cardiac function. In our paper, we were able to identify Lin28a as an important regulator of the heart muscle cells’ proliferation. This allows us to improve the outcome and cardiac function post-heart attack in a mouse model.
This new gene may be an answer to unmet problems that lead to ischemic heart disease, which is the leading cause of death for both men and women. This work emphasizes that selective modified mRNA, which allows transient but high gene expression in the cardiac muscle cells, is a platform that holds great potential for developing treatment for ischemic heart disease.
In the future, we would like to evaluate if Lin28a, delivered as selective modified mRNA, may induce cardiac repair after heart attack in a large animal model (e.g., pig model). If successful, these results may lead to clinical trials that may overcome this life-threatening disease.
Gregorio Lab

Carol Gregorio, PhD, center, with, from left, Nishtha Desai, MSc; Darshini Desai, PhD; Manish Manoharan, MSc; Svetlana Minakhina, PhD; Fabrizzio F. Caro; and Ann Anu Kurian, MSc.
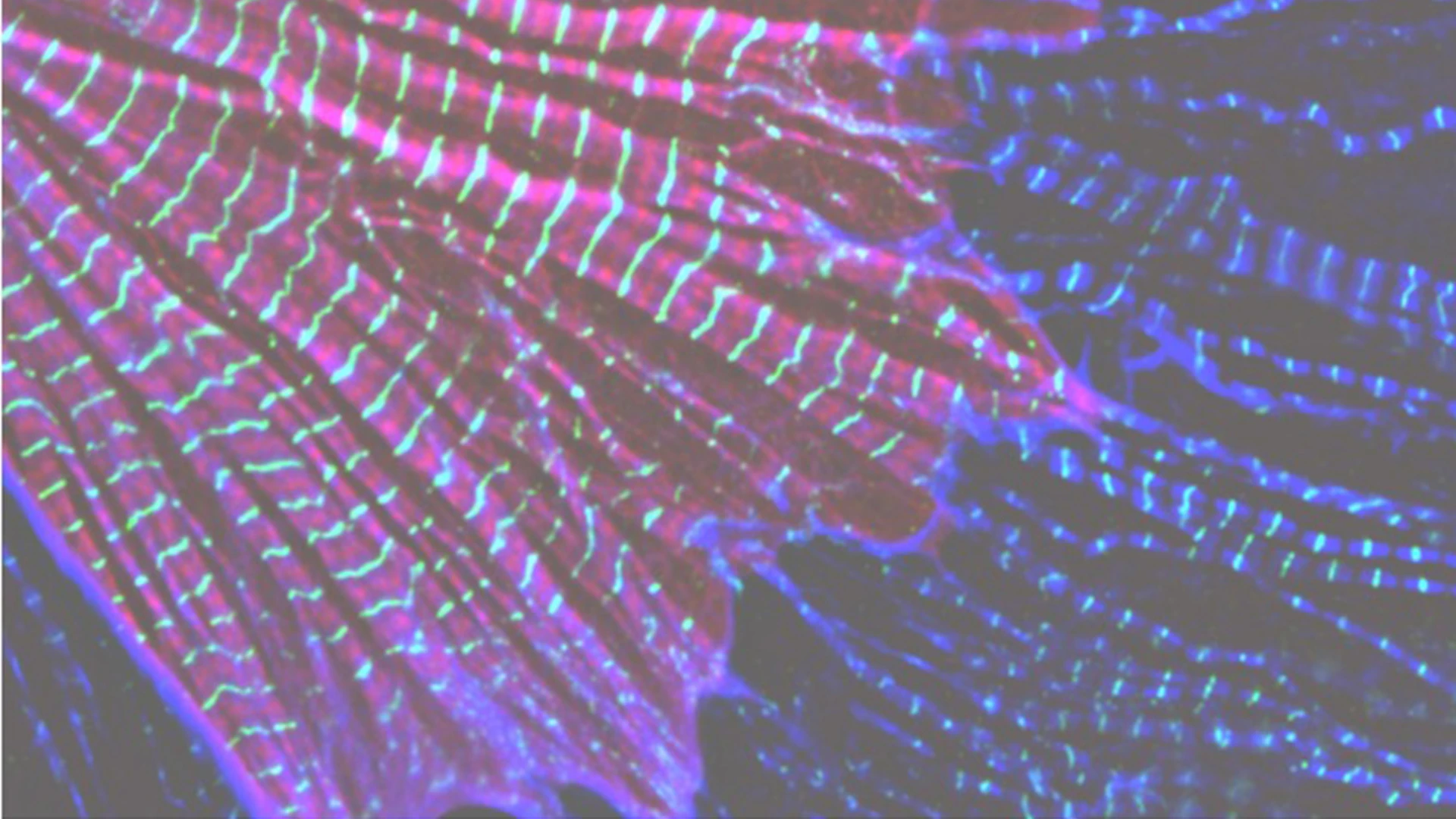
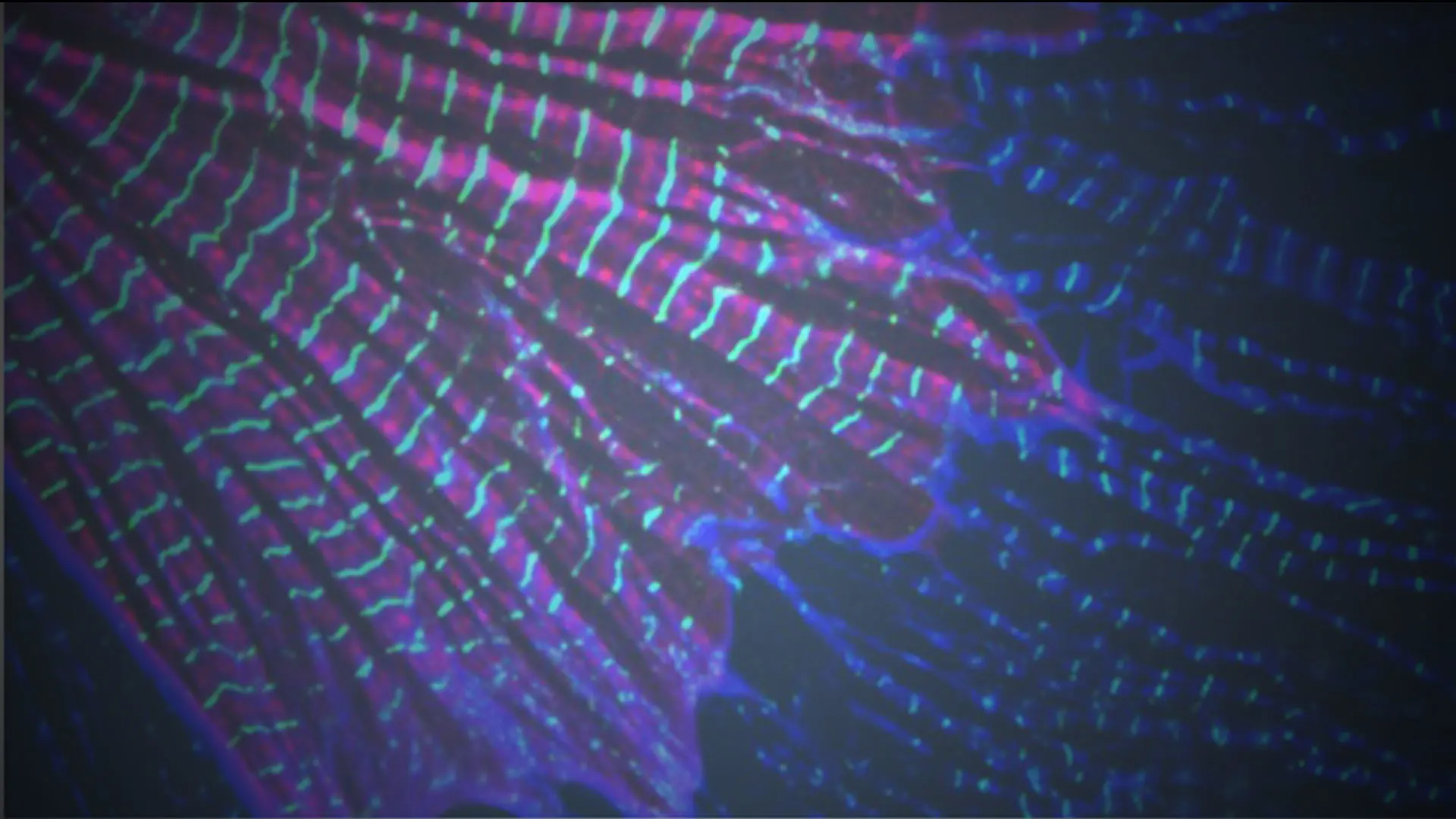
At the subcellular level, sarcomeres are individual units, essential for heart and skeletal muscle contraction. LMOD2 is required for maintaining thin filament lengths. Above is LMOD2 knockout cardiac myocyte, displaying disrupted sarcomere structure.
Abstract of Study: Human disease-causing mutations result in loss of leiomodin 2 through nonsense-mediated mRNA decay, PLOS Genetics, May 2024
The leiomodin (LMOD) family of actin-binding proteins plays a critical role in muscle function, highlighted by the fact that mutations in all three family members (LMOD1-3) result in human myopathies. Mutations in the cardiac predominant isoform, LMOD2, lead to severe neonatal dilated cardiomyopathy. Most of the disease-causing mutations in the LMOD gene family are nonsense, or frameshift, mutations predicted to result in expression of truncated proteins. However, in nearly all cases of disease, little to no LMOD protein is expressed. We show here that nonsense-mediated mRNA decay, a cellular mechanism that eliminates mRNAs with premature termination codons, underlies loss of mutant protein from two independent LMOD2 disease-causing mutations. Furthermore, we generated steric-blocking oligonucleotides that obstruct deposition of the exon junction complex, preventing nonsense-mediated mRNA decay of mutant LMOD2 transcripts, thereby restoring mutant protein expression. Our investigation lays the initial groundwork for potential therapeutic intervention in LMOD-linked myopathies.
Notes From the Investigator: Carol Gregorio, PhD, Professor of Medicine (Cardiology), and Cell, Developmental and Regenerative Biology
Contraction of striated muscle results from force generated by the sliding of actin-thin filaments past myosin-thick filaments. Therefore, precision of thin filament assembly is essential for muscle function, which is supported by the fact that alterations in thin filament length are associated with life-threatening diseases such as dilated cardiomyopathy (DCM) and nemaline myopathy. LMODs are actin-binding proteins that regulate actin polymerization. Identifying how LMOD2 regulates thin filament lengths is important for understanding the pathogenesis of DCM, since we found that thin filament lengths are shorter in the hearts of both humans and mice that present with DCM (regardless of the cause). We and others have discovered human LMOD2 mutations that lead to DCM, which are lethal within the first few weeks of life without intervention. We found that most mutations result in a near-complete loss of mutant LMOD2 protein expression due to nonsense-mediated mRNA decay (NMD). In this investigation led by Christopher Pappas, PhD, we determined that blocking NMD specific for mutant LMOD2 restores mutant protein expression, which we found is partially functional, improves cardiomyocyte function, and thus, could have therapeutic potential.